Page Not Found
Page not found. Your pixels are in another canvas.

A list of all the posts and pages found on the site. For you robots out there is an XML version available for digesting as well.
Page not found. Your pixels are in another canvas.
This is a page not in th emain menu
Published:
This bit of code can be a great help to subset a Fasta file alignment based on certain conditions taken from a sample information file (e.g. subset only the samples collected in Spring, or those in Winter and greater than y cm, or everything except those in Autumn, etc.).
Published:
This post will show you how to generate a phylogeny plot in R, with a heatmap alongside it. This example uses binary data, but it can be adapted to be plotted with a continuous variable.
Published:
This R code shows you how to plot a map for a desired country or geographic region, and how to display a chosen climatic variable (e.g. mean annual temperature, annual precipitation) as an overlay. The WorldClim dataset has 19 variables to choose from.
Published:
This R script plots the map of a desired country, and colours it by elevation. It then adds GPS coordinate points to it (for example sampling sites) from an Excel file. Check out the final image at the bottom of this post 😃. The North arrow and Tetramesa wasp was added manually later in Inkscape.
Published:
I came across the problem of renaming sequences in a FASTA sequence alignment after downloading over 200 sequences from GenBank for four different genes. The sequence names were assigned by the GenBank accession number (e.g. MK1526) only. I wanted the sequence names to have the species name in it as well, for example MK1526_Canis_lupis. To avoid the tedious task of manually doing this, I wrote a few lines of R code that I hope will be of use to others with the same issue.
Published:
Perhaps a published paper lists their GenBank accession numbers as a range; for example KC664779 - KC665461. In R, one can use the following code to download all the sequences in this range, and save them as a FASTA file:
Published:
This snippet of Python or R code (whichever you prefer!) enables you to input a folder directory containing many FASTA files containing a single sequence in each, and combine them all into one FASTA file. I initially found it cumbersome to manually copy and paste the FASTA sequence from each trimmed chromatogram file into one big FASTA file. This automates the process, and could be useful to folks who are generating FASTA files for alignments 😃. This works correctly in Python 3.9. If you’re more of an R person, the R script is here too 😎.
An R Shiny application for processing binary data from fragment analysis methods such as ISSR and AFLP. 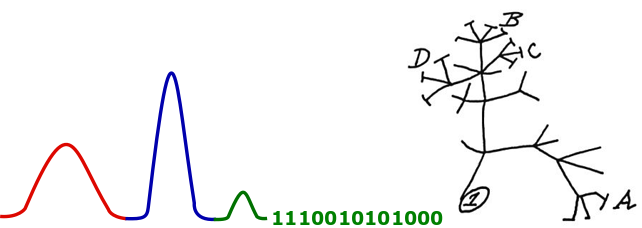
An R Shiny application to identify cochineal genetic sequences for 12S rRNA, 18S rRNA, or COI.
An R Shiny App that assesses the effects of methodological choice (e.g. tree prior, rate distribution, or clock rate) and sampling effects on the GMYC model for species delimitation.
Assesses sample size requirements for researchers performing critical thermal limits (CTL) studies
Published in Skeptical Inquirer, 2020
This article critically examines the claims in the book “Becoming Supernatural” by Joe Dispenza. 📁 PDF
Recommended citation: van Steenderen, C.J.M. 2020. Becoming Supernatural: How common people are being misled. Skeptical Inquirer Vol.44 No. 4. https://skepticalinquirer.org/authors/clarke-van-steenderen/
Published in Biological Control, 2021
Invasive Cactaceae cause considerable damage to ecosystem function and agricultural practices around the world. The most successful biological control agents used to combat this group of weeds belong to the genus Dactylopius (Hemiptera: Dactylopiidae), commonly known as ‘cochineal’. Effective control relies on selecting the correct species, or in some cases, the most effective intraspecific lineage, of cochineal for the target cactus species. Many of the Dactylopius species are so morphologically similar, and in the case of intraspecific lineages, identical, that numerous misidentifications have been made in the past. These errors have resulted in failed attempts at the biological control of some cactus species. This study aimed to generate a multi-locus genetic database to enable the accurate identification of dactylopiids. Genetic characterization was achieved through the nucleotide sequencing of three gene regions (12S rRNA, 18S rRNA, and COI) and two inter-simple sequence repeats (ISSR). Nucleotide sequences were very effective for species-level and D. tomentosus lineage-level identification, but could not distinguish between the two lineages within D. opuntiae commonly used for biological control of various Opuntia spp. Fragment analysis through the use of ISSRs successfully addressed this issue. This is the first time that a method has been developed that can distinguish between these two D. opuntiae lineages. Using the methods developed in this study, biological control practitioners can ensure that the most effective agent species and lineages are used for each cactus target weed, thus maximizing the level of control. 📁 PDF
Recommended citation: van Steenderen, C.J.M., Paterson, I.D., Edwards, S., and Day, M.D. 2021. Addressing the red flags in cochineal identification: The use of molecular techniques to identify cochineal insects that are used as biological control agents for invasive alien cacti. Biological Control 104426. doi: 10.1016/j.biocontrol.2020.104426. https://www.sciencedirect.com/science/article/pii/S1049964420306538
Published in Molecular Ecology Resources, 2022
Species delimitation tools are vital to taxonomy and the discovery of new species. These tools can make use of genetic data to estimate species boundaries, where one of the most widely-used methods is the Generalised Mixed Yule Coalescent (GMYC) model. Despite its popularity, a number of factors are known to influence the performance and resulting inferences of the GMYC. Moreover, the few studies that have assessed model performance to date have been predominantly based on simulated datasets, where model assumptions are not violated. Here, we present a user-friendly R Shiny application, “SPEDE-sampler” (SPEcies DElimitation sampler), that assesses the effect of computational and methodological choices, in combination with sampling effects, on the GMYC model. Output phylogenies are used to test the effect that 1) sample size, 2) BEAST and GMYC parameters (e.g. prior settings, single vs multiple threshold, clock model), and 3) singletons has on GMYC output. Optional predefined grouping information (e.g. morphospecies/ecotypes) can be uploaded in order to compare it to GMYC species and estimate percentage match scores. Additionally, predefined groups that contribute to inflated species richness estimates are identified by SPEDE-sampler, allowing for the further investigation of potential cryptic species or geographic sub-structuring in those groups. Merging by the GMYC is also recorded to identify where traditional taxonomy has overestimated species numbers. Four worked examples are provided to illustrate the functionality of the program’s workflow, and the variation that can arise when applying the GMYC model to empirical datasets. The R Shiny program is available for download on GitHub. 📁 PDF
Recommended citation: van Steenderen, C.J.M. and Sutton, G.F. 2022. SPEDE-sampler: an R Shiny application to assess how methodological choices and taxon-sampling can affect Generalised Mixed Yule Coalescent (GMYC) output and interpretation. Molecular Ecology Resources (22)2 doi: 10.1111/1755-0998.13591 https://onlinelibrary.wiley.com/doi/abs/10.1111/1755-0998.13591
Published in Biodiversity Data Journal, 2022
Processing and visualising trends in the binary data (presence or absence of electropherogram peaks), obtained from fragment analysis methods in molecular biology, can be a time-consuming and often cumbersome process. Scoring and analysing binary data (from methods, such as AFLPs, ISSRs and RFLPs) entail complex workflows that require a high level of computational and bioinformatic skills. The application presented here (BinMat) is a free, open-source and user-friendly R Shiny programme (https:// clarkevansteenderen.shinyapps.io/BINMAT/) that automates the analysis pipeline on one platform. It is also available as an R package on the Comprehensive R Archive Network (CRAN) (https://cran.r-project.org/web/packages/BinMat/index.html). BinMat consolidates replicate sample pairs of binary data into consensus reads, produces summary statistics and allows the user to visualise their data as ordination plots and clustering trees without having to use multiple programmes and input files or rely on previous programming experience. 📁 PDF
Recommended citation: van Steenderen, C.J.M. 2022. Biodiversity Data Journal (10) doi: 10.3897/BDJ.10.e77875 https://bdj.pensoft.net/article/77875/
Published in Bioinvasions Records, 2023
An undescribed phytophagous wasp belonging to the Tetramesa genus (Hymenoptera: Eurytomidae), that is native to South Africa, is currently being investigated as a potential weed biological control agent for the invasive grass Eragrostis curvula (Poaceae) in Australia. Host-specificity testing is underway in South Africa, but the wasp has not been exported into quarantine in Australia and further research is required before it could be considered for release. Here, we used DNA barcoding to demonstrate that Tetramesa specimens collected on invasive E. curvula populations in Australia represent the same wasp species currently being investigated in South Africa. We discuss our findings in the context of developing a biological control programme against E. curvula in Australia and the potential risk posed to native Australian grasses
Recommended citation: Sutton, G.F., van Steenderen, C.J.M., Yell, L.D., Canavan, K., McConnachie, A., and Paterson, I.D. 2023. First record of an African grass-feeding wasp (Tetramesa; Eurytomidae) on the invasive grass Eragrostis curvula (African lovegrass; Poaceae) in Australia. Bioinvasions Records 12(3):673-680 doi: 10.3391/bir.2023.12.3.05 https://www.reabic.net/journals/bir/2023/3/BIR_2023_Sutton_etal.pdf
Published in Physiological Entomology, 2023
Required sample sizes for a study need to be carefully assessed to account for logistics, cost, ethics and statistical rigour. For example, many studies have shown that methodological variations can impact the critical thermal limits (CTLs) recorded for a species, although studies on the impact of sample size on these measures are lacking. Here, we present ThermalSampleR; an R CRAN package and Shiny application that can assist researchers in determining when adequate sample sizes have been reached for their data. The method is particularly useful because it is not taxon specific. The Shiny application offers a user-friendly interface equivalent to the package for users not familiar with R programming. ThermalSampleR is accompanied by an in-built example dataset, which we use to guide the user through the workflow with a fully worked tutorial.
Recommended citation: van Steenderen, C.J.M., Owen, C.A., Sutton, G.F., Martin, G.D., Coetzee, J.A. 2023. Sample size assessments for thermal physiology studies: An R package and R Shiny GUI. Physiological Entomology doi: 10.1111/phen.12416 https://resjournals.onlinelibrary.wiley.com/doi/10.1111/phen.12416
Published in Journal of Natural History, 2023
The butterfly genus Pseudonympha and several related genera are endemic to southern Africa. Although many of the species are montane, some inhabit the arid interior of South Africa, offering an opportunity to study the palaeobiogeography of this biome. Morphological data (for all species of Pseudonympha and allied African and Asian genera) and molecular data (WG and COI genes for nine of the 15 species of Pseudonympha and all of the southern African endemic genera of Ypthimina) were compiled. Phylogenetic analysis indicated that Pseudonympha apparently originated in the Cape Fold Mountains about 15 Mya ago and spread steadily eastwards and northwards along the Great Escarpment during the aridification of the region, perhaps assisted by orogeny in the east and oceanic cooling in the west. Aridification cycles seem to have intermittently isolated some early lineages in elevated habitats in the interior, so that those lineages show lower speciation rates (or perhaps higher extinction rates) than those in the east. Four species delineation techniques indicated that some species are taxonomically oversplit. Based on genetic polyphyly and morphological similarity, we propose that the status of P. swanepoeli be reduced to that of a subspecies of P. varii, such that all the north-eastern populations from Harrismith to Tzaneen fall under P. varii swanepoeli van Son stat. n., and all the southern populations fall under P. varii varii van Son stat. n. Ultimately, the diversification of both of these lineages seems tied to their host plants’ response to aridification brought on by continental drift and orogeny. Sympatric organisms (eg cicadas) with biologies focused around different resources (eg savanna trees) show other patterns of diversification. The phylogenetic analysis of the subtribe Ypthimina also supports the monophyly of Paternympha, paraphyly of Ypthima, recognition of Thymipa Moore stat. rev. as a phylogenetic independent genus, and new relationships for Strabena.
Recommended citation: van Steenderen, C.J.M., Pringle, E.L., and Villet, M.H. 2023. Historical diversification of Pseudonympha Wallengren, 1857 (Lepidoptera: Nymphalidae: Satyrinae). Journal of Natural History (10) doi: 10.1080/00222933.2023.2257373 https://doi.org/10.1080/00222933.2023.2257373
Published in BioControl, 2023
Many native South African grass species have become invasive elsewhere in the world. The application of biological control to invasive grasses has been approached with trepidation in the past, primarily due to concerns of a perceived lack of host specific herbivores. This has changed in recent times, and grasses are now considered suitable candidates. The Tetramesa Walker genus (Hymenoptera: Eurytomidae) has been found to contain species that are largely host specific to a particular grass species, or complex of closely related congeners. Very little taxonomic work exists for Tetramesa in the southern hemisphere, and the lack of morphological variability between many Tetramesa species has made identification difficult. This limits the ability to assess the genus for potential biological control agents. Species delimitation analyses indicated 16 putative novel southern African Tetramesa taxa. Ten of these were putative Tetramesa associated with Eragrostis curvula (Schrad.) Nees and Sporobolus pyramidalis Beauv. and S. natalensis Steud., which are alien invasive weeds in Australia. Of these ten Tetramesa taxa, eight were only found on a single host plant, while two taxa were associated with multiple species in a single grass genus. The Tetramesa spp. on S. pyramidalis and S. africanus were deemed suitably host-specific to be used as biological control agents. Field host range data for the Tetramesa species on E. curvula revealed that the wasp may not be suitably host specific for use as a biological control agent. However, further host specificity testing on non-target native Australian species is required.
Recommended citation: van Steenderen, C.J.M., Sutton, G.F., Yell, L.D., Canavan, K., McConnachie, A., Day, M.D., and Paterson, I.D. 2023. Phylogenetic analyses reveal multiple new stem-boring Tetramesa (Hymenoptera: Eurytomidae) taxa: implications for the biological control of invasive African grasses BioControl (10) doi: 10.1007/s10526-023-10231-4 https://link.springer.com/article/10.1007/s10526-023-10231-4
Published in Biocontrol Science and Technology, 2024
African lovegrass, Eragrostis curvula (Schrad.) Nees (Poaceae), is a perennial grass native to southern Africa that has become problematic in many countries, most notably in Australia. A biological control programme against E. curvula in Australia has been initiated to mitigate the plant’s negative impacts. We present field-based host-specificity observations of natural enemies in the native distribution in South Africa and validate these observations with laboratory based no-choice host-specificity tests. Only three insect species were consistently found utilising E. curvula, namely two stem-galling wasps belonging to the genus Tetramesa (Hymenoptera: Eurytomidae), and a shoot-galling fly (Diptera: Chloropidae). The shoot-galling fly was found to utilise grasses from multiple non-target grass genera and was thus rejected. Both Tetramesa species were recorded on multiple native African Eragrostis species during field host-range surveys, and this relatively broad host range was confirmed by both wasp species completing development on several native Eragrostis species under no-choice conditions in the laboratory. At least three native Australian species (E. parviflora, E. trachycarpa, E. leptocarpa) are predicted to be suitable hosts for both Tetramesa species. Neither Tetramesa species are therefore likely to be suitably host specific for release on E. curvula in Australia. The recent discovery of an adventive population of one of the South African Tetramesa on E. curvula in Australia provides an opportunity to test whether the predicted host range of the species is realised in the introduced distribution.
Recommended citation: Yell, L.D., Sutton, G.F., van Steenderen, C.J.M., Canavan, K., McConnachie, A., and Paterson, I.D. 2024. Biocontrol Science and Technology (10) doi: 10.1080/09583157.2024.2317135 https://www.tandfonline.com/doi/full/10.1080/09583157.2024.2317135#abstract
Published in Ecological Modelling, 2024
The performance and transferability of species distribution models (SDMs) depends on a number of ecological, biological, and methodological factors. There is a growing body of literature that explores how the choice of climate covariate combinations and model parameters can affect predictive performance, but relatively few that delve into covariate reduction methods and the optimisation of model parameters, and the resulting spatial and temporal transferability of those models. The present work used the citrus pest, Diaphorina citri Kuwayama (Hemiptera: Psyllidae), to illustrate how MaxEnt models trained on the insect’s native range in Asia varied in their predictions of climatic suitability across the introduced range when eight different covariate reduction methods were applied during model building. Additionally, it showed how model sensitivity varied across these different covariate combinations using three sets of independently validated occurrence points in the invaded range of the psyllid. Climatically suitable areas for D. citri differed by as much as two-fold between the best and worst-performing models in selected areas. Great care should be taken in the selection of the highest-performing predictor combinations and model parameter settings for SDMs, particularly in the case of invasive species where the assumption of environmental equilibrium is likely violated in the introduced range. Understanding how the predictive ability of SDMs can be influenced by the methodological choices made during the model building phase is vital to ensuring that ecological and invasion management programmes do not over- or underestimate climatic suitability and subsequent invasion risk.
Recommended citation: van Steenderen, C.J.M. and Sutton, G.F. 2024. Ecological Modelling (10) doi: 10.1016/j.ecolmodel.2024.110872 https://www.sciencedirect.com/science/article/pii/S0304380024002606?via%3Dihub
Published in African Entomology, 2024
The Asian Citrus Psyllid (ACP) (Diaphorina citri Kuwayama, 1908) (Hemiptera: Psyllidae) is a major citrus pest. The species has been introduced to West and East Africa, but has not yet spread to southern Africa, where it could have a devastating impact on citrus farming and livelihoods. A proactive response is key to mitigating the species’ impacts, particularly the ongoing monitoring of potential invasion routes and entry points into South Africa. Species distribution models (SDMs) were developed under current and future climates for ACP in Africa, and these models were used to (1) determine where the species likely poses a threat, (2) identify potential invasion routes into South Africa, and (3) assess how these factors will be affected under climate change. The SDMs indicated that there is an almost contiguous band of suitable climate along the east coast of Africa that joins the species’ current range in East Africa to South Africa, and under aggressive climate change a potential route of invasion through Namibia and Botswana. Much of South Africa is climatically suitable for the species, but under climate change, climatically suitable areas are likely to shift further inland. The spread of ACP into South Africa is unlikely to be prevented, but the outputs of the present models will inform monitoring activities and assist with preparations to respond to this predicted biological invasion.
Recommended citation: van Steenderen, C.J.M., Mauda, E.V., Kirkman, W., Faulkner, K.T., and Sutton, G.F. 2024. African Entomology (10) doi: 10.17159/2254-8854/2024/a18476 10.17159/2254-8854/2024/a18476
Published in Austral Entomology, 2025
Two new species of gall midges are described whose larvae feed on the small morphotype of Guinea grass Megathyrsus maximus in Africa. Arabukodiplosis basalis Kolesik, gen. et sp. nov. causes galls at the base of plant’s crown in Kenya and Arabukodiplosis vesicaria Kolesik, gen. et sp. nov. causes blister galls on the stems in South Africa and Kenya. Description of the morphology and the sequence of a fragment of the COI mitochondrial gene of the insects are provided. A new genus is erected to contain the two new species. Arabukodiplosis Kolesik, gen. nov. belongs to the supertribe Cecidomyiidi and its closest relative is Mitodiplosis Kieffer, 1914, an African genus containing a single species that induces stem thickening galls on pyp grass Ehrharta villosa (Poaceae) in South Africa. Like Mitodiplosis, Arabukodiplosis Kolesik, gen. nov. cannot be satisfactorily accommodated in any of the currently recognised tribes. The crown of the plant, where A. basalis Kolesik, gen. et sp. nov. forms galls, is where new tillers and shoots originate, so the galls develop where stems would usually form. Stems infested by A. vesicaria Kolesik, gen. et sp. nov. continue to develop above the galls, but the gall is expected to act as a resource sink, reducing the fitness of the host plant. Both species are possible candidates for biological control of M. maximus, which is a serious invasive alien pest outside of its native distribution.
Recommended citation: . Kolesik, P., Sutton, G.F., van Steenderen, C.J.M., Martin, D., Plowes, R. and Paterson, I.D. 2024. Austral Entomology (10) doi: https://onlinelibrary.wiley.com/doi/10.1111/aen.12719?af=R
Published in Austral Entomology, 2025
The genus Tetramesa Walker (Hymenoptera: Eurytomidae) comprises over 200 species of herbivorous wasps that feed exclusively on grasses. Recent field surveys in South Africa for grass biological control programs have uncovered a large diversity of potential Tetramesa on African grasses. Here, mitochondrial (cytochrome c oxidase I [COI]) and nuclear (28S) genetic sequences were used to compare the outputs of seven popular species delimitation methods and to guide the generation of consensus species boundaries for putative Tetramesa taxa and close relatives. Additionally, the nuclear region was used to run a dated analysis that applied a molecular clock rate. Consensus species delimitation results found 35 molecular operational taxonomic units (MOTUs) in the COI data and 21 MOTUs in the 28S data. Of the 35 COI MOTUs, there were 17 putative Tetramesa taxa (16 novel southern African taxa and 1 described Northern Hemisphere species, Tetramesa romana), 13 of which showed evidence of specialisation to a single host plant. Comparatively, of the twenty-one 28S MOTUs, there were 5 putative Tetramesa taxa (4 novel southern African taxa and 1 T. romana), all of which showed evidence of host specificity. The dated analysis suggested that the genus Tetramesa originated ~67.1 mya. There was evidence of rapid diversification in the Southern Hemisphere clades between 5 and 15 mya, which coincides with grassland expansions and climatic fluctuations in Africa at the time that may have driven host specialisation. The present results provide valuable insights into the diversity and broader scale evolutionary patterns in this Southern Hemisphere microhymenopteran group.
Recommended citation: van Steenderen, C.J.M., Suton, G.F., Yell, L.D., Canavan, K., and Paterson, I.D. 202. Austral Entomology (10) doi: 10.1111/aen.70006 https://doi.org/10.1111/aen.70006
Published in Ecology and Evolution, 2025
Invasive populations of the slenderleaf iceplant, Mesembryanthemum nodiflorum L., are problematic along the west coast of North America. The plant is hypothesised to originate from southern Africa, though it has established populations in North Africa and the Mediterranean. There is interest in initiating a biological control programme for the weed in its invaded range, but a clearer understanding of its invasion pathways and sources of origin is required in order to prioritise potential biological control agents. This study used both NextGen RADseq and fragment analysis ISSR techniques to uncover the population structure and genetic diversity of M. nodiflorum in its native, introduced and invaded ranges. The results supported a South African origin of the species based on a higher number of private alleles and overall genetic diversity. Our results suggest a bridgehead effect, where a secondary invasion to North America from Mediterranean populations took place, rather than a direct invasion from the native range in South Africa. The present results indicated that surveys for potential biocontrol agents for M. nodiflorum should be conducted in the native South African distribution, where the greatest diversity of specialist natural enemies is likely to be present.
Recommended citation: van Steenderen, C.J.M., Sandenbergh, E., Brookes, D., Moran, Patrick, and Paterson, I.D. 2025. Ecology and Evolution (10) doi: 10.1002/ece3.72262 https://doi.org/10.1002/ece3.72262
Published:
The ability to use genetic barcoding tools to distinguish between the species and intraspecific ‘biotypes’ within the Dactylopius genus (Hemiptera: Dactylopiidae) is highly beneficial to the biological control of cactaceous weeds in South Africa. The present study used DNA sequencing and ISSR fragment analysis methods to create a database of genetic barcodes for Dactylopius species found in the country, as well as from the native range, with a particular focus on the biotypes found within Dactylopius opuntiae. This has important applications in the mass rearing of pure insect cultures and the inoculation of the most effective biotype on target Cactaceae.
Published:
Invasive Cactaceae cause considerable damage to ecosystem function and agricultural practices around the world but some cacti are also important and valued crop species. The most successful biological control agents used to combat cactus weeds belong to the genus Dactylopius (Hemiptera: Dactylopiidae), commonly known as ‘cochineal’, but the worst pests of cactus crops are also members of this genus. Cochineal lineages used for biocontrol of cactus weeds are host specific and only certain species and lineages will feed on cactus crops, so cactus biocontrol can be safely implemented without harm to cactus agriculture. Many of the Dactylopius species are so morphologically similar, and in the case of intraspecific lineages, identical, that numerous misidentifications have been made in the past. These errors may result in cactus farmers incorrectly assuming that the biocontrol agent is damaging their crop. This study aimed to generate a multi-locus genetic database to enable the accurate identification of dactylopiids. This was achieved through the nucleotide sequencing of three gene regions (12S rRNA, 18S rRNA, and COI) and two inter-simple sequence repeats (ISSR). Nucleotide sequences were very effective for species-level and D. tomentosus lineage-level identification, but could not distinguish between the two lineages within D. opuntiae commonly used for biological control of various Opuntia spp. Fragment analysis through the use of ISSRs successfully addressed this issue. This is the first time that a method has been developed that can distinguish between these two D. opuntiae lineages. Using the methods developed here, one can distinguish between what is a potential pest, and what is a beneficial biological control agent.
Published:
The Tetramesa genus (Hymenoptera: Eurytomidae) comprises at least 200 species that feed exclusively on grasses. The highly host-specific behaviour of these wasps, and the damage that they can cause to their host plants, makes them ideal biological control agent candidates for invasive grasses. Very little is known about the Afrotropical Hymenoptera in general, and to date, almost all of the sampling effort in collecting and describing Tetramesa species has taken place in the Northern Hemisphere. Only four African species have been described; none of which are from South Africa. The Centre for Biological Control (CBC) at Rhodes University has been investigating biological control options for several African grasses that have become invasive in Australia and the Americas, and have been collecting Tetramesa specimens across South Africa since 2017. The insect communities associated with more than 55 different native grasses have been surveyed over this period. The uniform morphology of adult and larval Tetramesa has, however, made it impossible to determine whether these wasps are a single polyphagous species, or multiple oligophagous and/or monophagous species. We are currently using genetic barcoding tools (mitochondrial COI and nuclear ITS2 regions) and species delimitation methods to solve this problem. Our preliminary results have identified at least four putative species (or rather ‘molecular operational taxonomic units’ (MOTUs)). These were collected from single host plants, confirming their host-specificity and potential as biological control agents. It is likely that we will uncover many more undescribed species in the region as our sampling effort escalates.
Published:
The stem-boring wasp genus Tetramesa (Hymenoptera: Eurytomidae) comprises 203 species that feed exclusively on grasses. The wasps are highly host-specific, typically feeding on a single or a few closely-related grasses, and can cause significant damage to their host grass (e.g. reducing seed production, increasing tiller mortality). These attributes often result in Tetramesa being serious grain pests, but it also makes them ideal biological control agent candidates for controlling invasive grasses. Very little is known about the Afrotropical Hymenoptera in general, and to date, almost all the sampling effort in collecting and describing Tetramesa species has taken place in the northern hemisphere. Only four African species have been described; none of which are from South Africa. The Centre for Biological Control (CBC) at Rhodes University has been investigating biological control options for several African grasses that have become invasive in Australia and the Americas, and have been collecting Tetramesa specimens across South Africa since 2017. The insect communities associated with more than 60 different native grasses have been surveyed over this period. The uniform morphology of adult and larval Tetramesa has, however, made it impossible to determine whether these wasps are a single polyphagous species, or multiple oligophagous and/or monophagous species. We are currently using genetic barcoding tools (mitochondrial COI and nuclear ITS2 regions) and species delimitation methods to solve this problem. Our preliminary results have identified at least six potentially undescribed Tetramesa species from South Africa. Each novel Tetramesa species was highly specific, with five of the six potential species feeding and completing their development on a single host grass species. This work will facilitate using biological control techniques to manage invasive alien grass species and highlights a previously unknown diversity of Tetramesa species associated with South African grasses. It is likely that we will uncover many more undescribed Tetramesa species in the region as our sampling effort escalates.







